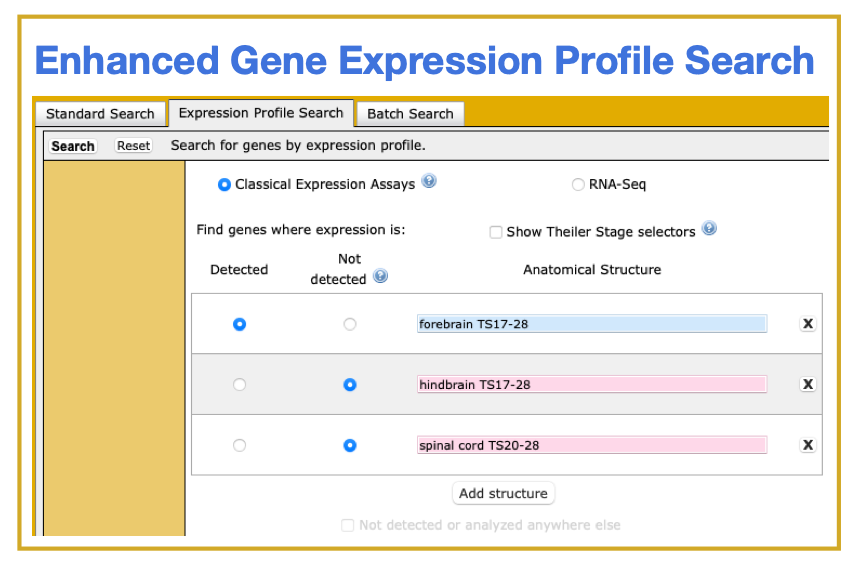
- Search
- Download
- More Resources
- More Resources Index
- Research Community E-mail Lists
- Online Books
- Nomenclature Home Page
- MGI Glossary
- Mouse Phenome Database (MPD)
- Deltagen and Lexicon Knockout Mice
- International Mouse Phenotyping Consortium (IMPC)
- Mouse Resources For COVID-19 Research
- Contributed Data Sets
- Community Links
- Software Developer Tools
- Submit Data
- Find Mice (IMSR)
 Analysis Tools
Analysis Tools- Contact Us
- Browsers