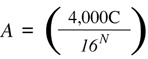
Without a doubt, the polymerase chain reaction (PCR) represents the single most important technique in the field of molecular biology today. What PCR accomplishes in technical terms can be described very simply — it allows the rapid and unlimited amplification of specific nucleic acid sequences that may be present at very low concentrations in very complex mixtures. Within less than a decade after its initial development, it has become a critical tool for all practicing molecular biologists, and it has served to bring molecular biology into the practice of many other fields in the biomedical sciences and beyond. The reasons are several fold. First, PCR provides the ultimate in sensitivity — single DNA molecules can be detected and analyzed for sequence content (Li et al., 1988; Arnheim et al., 1990). Second, it provides the ultimate in resolution — all polymorphisms, from single base changes to large rearrangements, can be distinguished by an appropriate PCR-based assay. Third, it is extremely rapid — for many applications, it is possible to go from crude tissue samples to results within the confines of a single workday. Finally, the technique is an agent of democracy — once the sequences of the pair of oligonucleotides that define a particular PCR reaction are published, anyone anywhere with the funds to buy the oligonucleotides can reproduce the same reaction on samples of his or her choosing; this stands in contrast to RFLP analyses in which investigators are often dependent upon the generosity of others to provide clones to be used as probes. Numerous books and thousands of journal articles have been published on the principles and applications of the technique [Erlich (1989) and Innis et al. (1990) are two early examples].
Although the applications of PCR are as varied as the laboratories in which the technique is practiced, this section will focus entirely on six general applications that are relevant to the detection and typing of genetic variation in the mouse. Four of these applications are based on the PCR amplification of particular loci that have been previously characterized at the sequence level. In these cases, primer pairs must be chosen to be as specific as possible for the locus in question in order to avoid artifactual PCR products. Computer programs are available to assist in primer design (Lowe et al., 1990; Dietrich et al., 1992), but manual inspection is usually adequate. One must be careful to avoid self-complementarity within any one primer and the presence of complementary sequences between the two primers. Also, potential primers should be screened with use of a sequence comparison program to avoid homology with the highly repeated elements B1, B2, and L1 (see Section 5.4). The primer length should be at least 20 bases, the G:C content should be at least 50%, and the melting temperature should be at least 60°C.
Even when all of these conditions are adhered to, it is still possible to find that a particular pair of primers will not work properly to amplify a specific locus into a reproducible product that can be clearly distinguished from artifactual background bands. There are a variety of approaches that one can take to eliminate such problems (Erlich, 1989; Innis et al., 1990), but if all else fails, one should replace one or both primers with alternatives derived from other nearby flanking sequences that also fit the rules listed above.
Rapid, highly efficient PCR-based assays can be designed to detect all RFLPs — previously-defined by Southern blot analysis — as long as the nature of the RFLP is understood and sequence information flanking the actual polymorphic site is available. A pair of PCR primers that flank this site can then be synthesized according to the rules just described and tested for their ability to amplify a specific product that can be readily identified as an ethidium bromide-stained band by gel electrophoresis.
With the simplest and most common type of RFLP illustrated in Figure 8.6, the polymorphism results from a single nucleotide difference that provides a recognition site for a restriction enzyme in one allelic form and not the other. A polymorphism of this type can be rapidly detected by (1) amplifying the region around the polymorphic site from each sample; (2) subjecting the amplified material to the appropriate restriction enzyme for a brief period of digestion; and (3) distinguishing the undigested PCR product from the smaller digested fragments by gel electrophoresis. By choosing primers that are relatively equidistant to and sufficiently far from the polymorphic site, one can easily resolve allelic forms on agarose or polyacrylamide gels as illustrated in Figure 8.6.
This PCR-based protocol provides results much more rapidly and is much easier to carry out than the Southern blot alternative which requires blotting, probe labeling, hybridization, and autoradiography. Since the major expense involved with PCR is in the initial sequencing of the locus and the synthesis of PCR primers, it is also less costly in all cases where one expects to type large numbers of samples for the particular locus in question.
Even RFLPs caused by more complex mutational events can be analyzed by PCR. Figure 8.7 illustrates the logic behind devising PCR strategies for detecting deletions, insertions, inversions, and translocations. The only requirement is a knowledge of the sequences that surround the breakpoints associated with each particular genetic event.
An important resource for the identification of new restriction site polymorphisms that can be typed by PCR is the 3'-untranslated (3'-UT) regions of transcripts. These regions are not under the same selective constraints as coding sequences and are frequently just as polymorphic as random non-transcribed genomic regions. However, 3'-UT regions are direct markers for the 3' ends of genes. They are usually not interrupted by introns and are often sufficiently divergent between different members of a gene family to allow locus-specific analysis.
Nearly all cDNA libraries are constructed from cDNA molecules that have been initiated by priming from the poly(A) tail present at the 3' end of the mRNA. For all clones obtained from these libraries, it is straightforward to obtain sequence information for a few hundred basepairs of 3'-UT region directly adjacent to the poly(A) tail. This sequence information can be used to design a pair of PCR primers that can be used, in turn, to amplify and sequence the same region from a different strain or species of mice such as M. spretus or M. m. castaneus. In a comparison of 2,312 bp present in 3'-UT regions derived from 22 random mouse cDNA clones, Takahashi and Ko (1993) found an overall polymorphism rate of one change in every 92 bp. These single base changes translated into restriction site polymorphisms within nine of the 22 clones analyzed. With primers already in-hand, these newly identified polymorphisms provide PCR markers for the direct mapping of corresponding genes that are indistinguishable in their coding regions.
Although PCR detection of RFLPs is an improvement over Southern blot detection, the real advantage of PCR lies within its nearly universal ability to discriminate alleles differing by single base changes even when they do not create or destroy any known restriction site. In fact, most random basepair changes will be of the non-RFLP type, and before the advent of PCR, there was no efficient means by which these alleles could be easily followed in large numbers of samples. It was this limitation that led originally to the development of the PCR protocol (Saiki et al., 1986).
The inability to detect single base changes on Southern blots was a consequence of both theoretical limitations inherent in the process of hybridization as well as practical limitations in the sensitivity of nucleic acid probes and the elimination of background noise. With Southern blot analysis, the sensitivity at which target sequences can be detected within a defined sample is directly proportional to the length of the probe. For example, a 1 kb probe will hybridize to ten times the amount of target sequence as a 100 bp probe (having the same specific activity), and this will lead to a signal which is ten times stronger. It is for this reason that it is always best to use the longest probes possible for traditional Southern blot studies as well as for other protocols such as in situ hybridization. Signal strength is important not simply to reduce the amount of time required for autoradiographic exposure, but also to allow detection over the background "noise" inherent in any hybridization experiment. If conditions are at all less than optimal, the signal to noise ratio will drop below 1.0 as the probe size is reduced below 100-200 bp.
The only forces holding the two strands of a DNA double helix together are the double or triple hydrogen bonds that exist within each basepair. Individual hydrogen bonds are very weak, and it is only when they are added together in large numbers that the double helix has sufficient stability to avoid being split apart by normal thermal fluctuations. Thus, for DNA molecules having a size in the range from a few basepairs up until a critical value of ~50 bp, the length itself plays a critical role in the determination of whether the helix will remain intact or fall apart. However, once this upper boundary is crossed, length is no longer a factor in thermal stability. In effect, there is only a small window — ~10 to 40 bp — over which it is possible to obtain differential hybridization of probe to target based on differences in hybrid length.
But how could length make a difference in allele detection when both the target and probe lengths are held constant? The answer is that the effective length of a hybrid is determined by the longest stretch of DNA that does not contain any mismatches. Thus, when a probe of 21 bases in length hybridizes to a target that differs at a single base directly in the middle of the sequence, the effective length of the hybrids that are formed is only 10 bp. Since a 10 bp hybrid is significantly less stable than a 21 bp hybrid, it is becomes easy to devise hybridization conditions (essentially by choosing the right temperature) such that the perfect hybrid will remain intact while the imperfect hybrid will not. In contrast, the thermal stability of a 50 bp hybrid is not sufficiently different from the thermal stability of a 100 bp hybrid (of equivalent sequence composition) to allow detection by differential hybridization.
Thus, in 1985, the detection of single base differences through differential Southern blot hybridization was not possible because of two counteracting problems. First, it was only with very short probes — oligonucleotides of less than 50 bases — that single base changes provided a large enough difference to be readily detected. However, it was only with much longer probes — of several hundred bases or greater — that signal strength and signal to noise ratio were sufficient to allow specific detection of the target sequence in any allelic form within the high complexity mouse genome. How could one break this impasse?
The answer, of course, was to focus on the target sequences rather than the probe or hybridization conditions. PCR provided a means to increase the absolute amount of target sequence, as well as the target to non target ratio, by virtually-unlimited orders of magnitude. This, in turn, results in a proportional increase in potential signal strength which, in turn, allows one to use short oligonucleotides for hybridization and which, in turn, allows for the detection of single base differences in a simple plus/minus assay.
Once alternative alleles have been sequenced and a single basepair change between the two has been identified, it becomes possible to design a PCR protocol that allows one to follow their segregation (Farr, 1991). First, PCR primers are identified that allow specific amplification of a region that encompasses the variant nucleotide site (for this application, the length of the product is not critical and can be anywhere from 150 to 400 bp in length). Next, two allele-specific oligonucleotides (ASOs) are produced in which the variant nucleotide is as close to the center as possible considering other factors described at the front of Section 8.3. The ideal ASO length is 19-21 bases — short enough to allow differential hybridization based on a single base change and long enough to provide a high probability of locus specificity. The two ASOs are used with defined samples to determine a temperature at which positive hybridization is obtained with target DNA containing the correct allele but not with target DNA containing only the alternative allele.
Typically, a sample of genomic DNA is subjected to PCR amplification with the locus-specific primers, aliquots of the amplified material are spotted into two "dots," and each is probed with labeled forms of each of the two ASOs. Hybridization at one dot but not the other is indicative of a homozygote for that allele, while hybridization at both dots is indicative of a heterozygote that carries both alleles.
The power of this protocol for allele detection is its simplicity. The elimination of gel running saves both time and allows for easy automation. With large amounts of target sequence, it becomes possible to use non-radioactive labeling protocols that are safer and allow for long-term storage of labeled probes (Helmuth, 1990; Levenson and Chang, 1990). However, there are pitfalls that are important to keep in mind. First, some variants may be refractory to reproducible PCR analysis because of problems inherent in the sequence that surrounds the site of the base change. Second, plus/minus assays of any kind are subject to the problem of false negatives. (One can always insert a gel running step prior to hybridization to be certain that amplified material is actually present in the aliquot under analysis.) Finally, in the analysis of mice derived from anything other than a defined cross, there is always the risk that a third novel allele will exist that cannot be detected by either of the two ASOs developed for the analysis. An animal heterozygous for such a novel allele (along with one of the two known alleles) could be falsely characterized as homozygote for the one known allele present since the protocol is not quantitative. Nevertheless, even with these pitfalls, the PCR/ASO protocol remains a useful tool for genetic analysis.
An alternative protocol for the detection of well-defined alleles that differ by single base changes has been developed by Hood and his colleagues (Landegren et al., 1988; Landegren et al., 1990). This method, called the oligonucleotide ligation assay (OLA) or ligase-mediated gene detection, is predicated on the requirement for proper base pairing at the 3'-end of one oligonucleotide as well as the 5'-end of an adjacent oligonucleotide before ligase can work to form a covalent phosphodiester bond. The conceptual framework behind the protocol is illustrated in Figure 8.8. First, the potential target sequence is amplified by PCR. Next hybridization is carried out simultaneously with two oligonucleotides complementary to sequences that are adjacent to each other and directly flank the variant nucleotide. The variant base itself will be either complementary or non-complementary to the most 3'-base of the first allele-specific oligonucleotide. This ASO is modified ahead of time with an attached biotin moiety but is not otherwise labeled. The second oligonucleotide, which is labeled radioactively or non-isotopically, extends across an adjacent sequence that is common to both alleles under analysis. Ligase is also present in the reaction, and if both oligonucleotides are perfectly matched with the target sequence, the ligase will create a covalent bond between them. If a mismatch occurs at the junction site, the two oligonucleotides will not become ligated. Biotinylated material can be easily and absolutely separated from non-biotinylated material with the use of a streptavidin matrix and the resulting sample can then be tested for the presence of the label associated with the second oligonucleotide.
There are two main advantages to using the ligase-mediated detection protocol as a substitute for the PCR/hybridization protocol described in the previous section. First, the chance of a false positive arising from the OLA protocol is essentially zero. Second, the OLA protocol is highly amenable to automation. However, as in all plus/minus assays, proper controls are critical to rule out the possibility of false negatives.
By combining OLA together with the exponential amplification strategy of PCR, a new technique has been developed that is referred to as the ligase chain reaction or LCR (Barany, 1991; Weiss, 1991). Like OLA, detection of nucleotide differences with the LCR protocol is based upon a requirement for perfect pairing at the two sites that flank the break between two oligonucleotides in order for ligase to form a phosphodiester bond between them as illustrated in Figure 8.8. The difference is that in the LCR protocol, four oligonucleotides are used corresponding to the regions that flank the polymorphic site on both strands of the target DNA molecule. Therein lies the mechanism of amplification. If the target sequence provides a match, both sets of flanking oligonucleotides will become ligated. After denaturation, each of the newly created double-length oligonucleotides can now act as a template for a new set of oligonucleotides to basepair and ligate. Thus, LCR proceeds by rounds of annealing in the presence of a heat-stable ligase followed by denaturation and then annealing again. The only difference in the thermocycler pattern from that used for PCR is the elimination of the elongation step. At the end of the process, the products of LCR can be detected easily in the same manner used for OLA as shown in Figure 8.8. In contrast, detection of polymorphic sites with PCR requires a follow-up protocol — either hybridization to an allele-specific oligonucleotide or restriction digestion followed by gel electrophoresis.
The essential difference between LCR and the original OLA protocol is sensitivity: LCR requires far less starting material since the product is amplified during the protocol. The advantages of the LCR protocol over PCR are several. First, like OLA, LCR will not produce false positives. Second, because the product of LCR is directly assayable without further detection schemes, the process is much more amenable to automation and is much more likely to be quantitative. The disadvantage of LCR is that it can only be used to detect single base substitutions that have been previously characterized by sequence analysis.
There are many circumstances where it is most useful to be able to follow genes — in contrast to anonymous sequences — directly within experimental crosses. A number of different approaches have already been described, but they all have limitations. Gene-associated RFLPs are detected between different species — M. spretus and traditional inbred M. musculus strains, for example — at a reasonable frequency, but are much more difficult to find among the inbred M. musculus strains themselves. To use any of the approaches dependent on Allele-Specific Oligonucleotides, it is first necessary to sequence the locus in question from different strains of mice, identify basepair variants, synthesize the ASOs as well as other locus-specific primer(s), test their specificity and optimize the reaction conditions for each locus. And at this point, one still only has a protocol for distinguishing two allelic states.
A geneticist's perfect protocol for detection and analysis of polymorphisms at any locus would satisfy the following criteria. First, it would allow the detection of any and all basepair variants in a DNA region as multiple alleles. Second, it would not require prior sequence information from each allele. Third, it would not require the synthesis of ASOs. Fourth, the assay protocol itself would require no special equipment or special skills above and beyond that found in a standard molecular biology laboratory. Finally, the assay would be rapid and the results would be readily reproducible.
All of these criteria have been satisfied, to a good degree, with a simple protocol that takes advantage of the fact that even single nucleotide changes can alter the three dimensional equilibrium conformation that single strands will assume at low temperatures (Orita et al., 1989a). If a sample of DNA is denatured at high temperature and then quickly placed onto ice, reformation of DNA hybrids will be inhibited. Instead, each single strand will collapse onto itself in what is often called a random coil. In fact, it is now clear that each single strand will assume a most-favored conformation based on the lowest free energy state. Presumably, the most favored state is one in which a large number of bases can form hydrogen bonds with each other. Even a single nucleotide change could conceivably disrupt the previous most favored state and promote a different one, which, if different enough, would run with an altered mobility on a gel. Different allelic states of a locus that are detected with this protocol are called single-strand conformation polymorphisms (SSCPs) (Beier et al., 1992; Beier, 1993).
The development of the SSCP protocol was an outgrowth of an earlier technique that allowed the detection of single base changes in genomic DNA upon electrophoresis through an increasing gradient of denaturant (Fischer and Lerman, 1983). This technique is called denaturing gradient gel electrophoresis (DGGE). Small changes in sequence have dramatic effects on the point in the denaturing gradient at which particular double-stranded genomic restriction fragments would split into single strands. With the attachment of a "GC-clamp" — composed of a stretch of tightly bonding G:C basepairs — the DNA fragment could be held together with a double helix in the clamped region attached to the open single strands present in the melted region. This two-phase molecule would be very resistant to further migration in the gel and would essentially stop in its track. Two allelic forms of a genomic fragment that differed by even a single basepair would undergo this transition at different denaturant points and this would be observed as different migration distances in the denaturing gradient gel. In the original protocol, different allelic forms were detected directly within total genomic DNA upon Southern blotting and hybridization to a locus-specific probe. At the time DGGE was developed, there was no other means available for detecting basepair changes that did not alter restriction sites, and thus DGGE expanded the polymorphism horizon. Unfortunately, DGGE requires the use of custom-made equipment and is tedious to perform on a routine basis.
In recent years, the DGGE protocol has been modified for use in conjunction with PCR as a means for the initial detection of allelic variants among samples recovered from different individuals within a population (Sheffield et al., 1989). The differential migration of PCR products can be detected directly in gels with ethidium bromide staining and variant alleles can be excised from the gel for sequence analysis. The main advantage to the use of DGGE is that nearly all single base substitutions can be detected (Myers et al., 1985). Thus, it is ideal for the situation where one wants to search for rare variant alleles among individuals within a population without the need for sequencing through all of the wild-type alleles that will be present in most samples. Nevertheless, DGGE does not scale up easily and, thus, it is not the method of choice when another less labor-intensive protocol can also be used for the detection of allelic variants. In many cases, the better protocol will be SSCP.
One of the main virtues of the new SSCP detection protocol developed by Orita et al. (1989a; 1989b) is its simplicity. In its basic form, PCR products are simply denatured at 94°, cooled to ice temperature to prevent hybrid formation, and then electrophoresed on standard non-denaturing polyacrylamide gels.
The two strands of the PCR product will usually run with very different mobilities, and base changes can act to further alter the mobility of each strand. Thus, what appears to be a single PCR product by standard analysis, can split into four different bands on an SSCP gel if the original DNA sample was heterozygous for base changes that altered the mobility of both strands.
Various studies have analyzed pairs of PCR products known to differ by single base substitutions to obtain an estimate of the fraction that can be distinguished by the SSCP protocol. In one such study, 80% of 228 variant PCR products were distinguishable (Sheffield et al., 1993). In another study, the rate of detection was 80-90% (Michaud et al., 1992). However, when this last set of samples was analyzed under three different electrophoretic conditions, the detection rate was an astonishing 100%.
When SSCP analysis was performed on a set of PCR products amplified from either the 3'-untranslated or intronic regions of 30 random mouse genes, 43% showed polymorphism in a comparison of the classical inbred strains B6 and DBA, and 86% showed polymorphism between B6 and M. spretus (Beier et al., 1992). The rate at which SSCP polymorphisms are detected between B6 and DBA is much greater than that observed with RFLP analysis, and in the same ballpark as the polymorphism frequencies observed for microsatellites described in Section 8.3.6.
There are a number of advantages to the SSCP approach over other systems for detecting polymorphisms: (1) SSCP has potential applicability to all unique sequences, both within genes and non-genic regions; (2) PCR primers can be designed directly from cDNA sequences; and (3) If one amplified region does not show polymorphism, one can always move upstream or downstream to another. However, it is still likely to be the case that microsatellites — described in Section 8.3.6 — will have larger numbers of different alleles and this makes them more useful in straightforward fingerprinting approaches. Together, SSCP and microsatellite analysis provide a powerful pair of PCR-based tools for classical linkage analysis with recombinant panels derived from both intra- and interspecific crosses.
With all of the PCR protocols described so far, there is an absolute requirement for pre-existing sequence information to design the primers upon which specific amplification depends. In 1990, two groups demonstrated that single short random oligonucleotides of arbitrary sequence could be used to prime the amplification of genomic sequences in a reproducible and polymorphic fashion (Welsh and McClelland, 1990; Welsh et al., 1991; Williams et al., 1990). This protocol is called random amplification of polymorphic DNA (RAPD). The principle behind the protocol is as follows. Short oligonucleotides of random sequence will, just by chance, be complementary to numerous sequences within the genome. If two complementary sequences are present on opposite strands of a genomic region in the correct orientation and within a close enough distance from each other, the DNA between them can become amplified by PCR. Each amplified fragment will be independent of all others and, by chance, will likely be of different length as well; if few enough bands are amplified, all will be resolvable from each other by gel electrophoresis. Different oligonucleotides will amplify completely different sets of loci.
RAPD polymorphisms result from the fact that a primer hybridization site in one genome that is altered at a single nucleotide in a second genome can lead to the elimination of a specific amplification product from that second genome as illustrated in Figure 8.9. If, for example, the random primer being used has a length of 10 bases, then each PCR product will be defined by 20 bases (10 in the primer target at each end) that are all susceptible to polymorphic changes. 60 The resulting polymorphism will be detected as a di-allelic +/ - system.
If one starts with the assumption that complete complementarity between primer and target is required for efficient amplification, it becomes possible to
derive a general equation to predict the approximate number, A, of amplified bands expected as PCR products from a genome of complexity C that
is primed with a single oligomer of length N.
61
For amplified fragments of 2 kb or smaller in size
62
the equation is:
Let us use 2x109 as an estimate for the complexity of the single copy portion of the mouse genome (see Section 5.1.2) and solve Equation 8.1 for primer lengths that vary from eight to 11. With N = 8, the predicted number of PCR products is 18,626 — far too many to resolve by any type of gel analysis. With N = 9, the equation predicts 116 PCR products, which is still too high a number. With N = 10, the prediction is 7.2 products, and with an 11-mer, the prediction is 0.45 products. Thus, the use of random 10-mers would be most appropriate for obtaining a maximal number of easily resolvable bands from the mouse genome.
Optimizations of the complete RAPD protocol, from the parameters upon which primer sequences are chosen to the conditions used for PCR, have been published (Williams et al., 1990; Nadeau et al., 1992). It is actually possible to obtain multiple PCR products with primers longer than 10-mers when relaxed reaction conditions are used to allow amplification from mismatched target sequences. In fact, one group has suggested that 12-14-mer primers are optimal (Nadeau et al., 1992). It is also possible to increase the predicted number of products by a simple factor of three by including two unrelated random primers of the same length in each PCR reaction. However, in any case where the number of visible PCR products goes above 12-20, it would become necessary to use polyacrylamide gels, rather than agarose gels, in order to clearly resolve each band; thus, the trade-off for the detection of more loci is a more time-consuming analysis. In the end, the protocol that requires the least amount of time for typing per locus is the one which should be chosen. Since different laboratories often excel at different techniques, the optimal conditions for RAPD analysis should be determined independently in each laboratory.
A comprehensive RAPD analysis of the two most well-characterized inbred strains — B6 and DBA — has been performed with 481 independent 10-mers used singly in PCR reactions (Woodward et al., 1992). An average of 5.8 PCR products per reaction were observed, which is not very different from that predicted from Equation 8.1. In a direct strain comparison, 95 reproducible differences were observed between B6 and DBA among the complete set of 2,900 discrete bands detected. Assuming that each polymorphism results from a single nucleotide change in one of the two primer targets and all such changes are detectable, it becomes possible to calculate the average sequence difference between these two strains at 1.6 changes per 1,000 nucleotides. This low level of polymorphism is not unexpected given the high degree of relatedness known to exist among all of the classical inbred strains (see Section 2.3.4).
Using RAPD as a method for detecting polymorphisms between B6 and DBA would appear to be rather inefficient — on average, only one polymorphism was detected among every five primer reactions that were run. A second negative factor is that all RAPD polymorphisms are binary +/ - systems. Thus, as discussed above for RFLPs, a polymorphism detected between one pair of strains may not translate into use for another pair of strains. Furthermore, it is not possible to distinguish animals that are heterozygous at any locus from those that are homozygous for the "+" allele. Thus, on average, only half of the RAPD polymorphisms detected between two strains would be mappable among offspring from a backcross to one parent, and with an intercross mapping system, the RAPD approach is even more limited (see Section 9.4.3).
Nevertheless, there are many features that speak to the utility of the RAPD approach. Foremost among these is the relative speed and ease with which results can be obtained — there is no need for blotting or radioactive hybridization, and a complete analysis from start to finish can be performed within a single working day, unlike RFLP or minisatellite studies. Unlike all other PCR-based protocols, RAPD primers are not dependent on the results of costly cloning and sequencing studies, and once they are obtained, the main cost per sample is the DNA polymerase used for PCR. Thus, even in comparisons of inbred strains, the RAPD protocol may be more efficient in the long run relative to other techniques for generating random DNA markers. Additionally, cloning of RAPD fragments can be rapidly accomplished after the simple recovery of ethidium bromide-detected bands. Cloned RAPD loci will have an advantage over minisatellites and microsatellites in that RAPD loci need not, and most will not, contain repetitive sequences.
As is the case with traditional RFLP loci, the interspecies level of RAPD polymorphism is much greater than that observed among the traditional inbred strains. The data of Serikawa et al. (1992) indicate a five-fold increase in the number of polymorphic bands observed in comparisons between M. spretus and traditional laboratory strains; this increase parallels the known increase in genetic diversity. Thus, the RAPD technology will be even more efficient for marker development in crosses that incorporate one parent that is not derived from one of the traditional inbred strains.
Like minisatellite analysis, the RAPD protocol can provide genomic fingerprints that simultaneously scan loci dispersed throughout the genome. In an analysis of 32 representative inbred strains maintained at the Jackson Laboratory, Nadeau and colleagues (1992) defined 29 unique strain fingerprints with the use of only six primers. Thus, RAPD would appear to provide an efficient and easy means by which to monitor the genetic purity of inbred lines on an ongoing basis.
Finally, it should be mentioned that while the RAPD protocol is a useful, and important, addition to the arsenal of tools available for genetic analysis of the mouse, it is of vastly greater importance for genetic studies of other species, including both animals and plants, that are not well-characterized at the DNA level. For these other species, the RAPD technology can provide a unique method for the rapid development of genetic markers and maps even before DNA libraries and clones are available.
The principle behind the RAPD approach is that oligonucleotides having essentially a random sequence will be present at random positions in the genome (of the mouse and every other species) at a frequency that can be predetermined mathematically. Thus, by choosing oligonucleotides of an appropriate size and by running PCR amplification reactions under the appropriate conditions, one can control the number of independent genomic fragments that are amplified such that they can be optimally resolved by a chosen system of gel electrophoresis. Although useful for linkage analysis, the RAPD approach does not allow the discrimination of mouse sequences from those of other species and, thus, it cannot be used as a means for recovering mouse genomic fragments from cells that contain a defined portion of the mouse genome within the context of heterologous genetic background.
An alternative approach that, like RAPD, also allows the simultaneous PCR amplification of multiple genomic fragments is based on the natural occurrence of highly repeated DNA elements that are dispersed throughout the genome. Three families of mouse repeat elements — B1, B2, and L1 — are each present in approximately 100,000 copies (see Section 5.4). Amplification of these elements in and of themselves with repeat-specific primers would not be useful, first, because their copy numbers are too great to be resolved by any available gel system and, second, because there would be no way of distinguishing most individual elements from each other since most would be of the same consensus size. However, if instead one used a repeat-specific primer that "faced-out" from the element, one would amplify only regions of DNA present between two elements that were sufficiently close to each and in the correct orientation to allow the PCR reaction to proceed. The number of instances in which two elements would satisfy these conditions will be much lower than the total copy number since, on average, these elements will be spaced apart at distances of approximately 30 kb 63 and, for all practical purposes, PCR amplification does not occur over distances greater than 1-2 kilobases. By working with (1) one or a combination of two or more primers together that (2) hybridize to whole families or subsets of elements within a family, from (3) two ends of the same element or from different elements, one can adjust the number of PCR products that will be generated to obtain the maximal number that can be resolved by a chosen system of gel electrophoresis (Herman et al., 1992).
This general protocol is referred to as Interspersed Repetitive Sequence (IRS) PCR (IRS-PCR). It was first developed for use with the highly repeated Alu family of elements in the human genome (Nelson et al., 1989), and was subsequently applied to the mouse genome (Cox et al., 1991). Cox and co-workers used individual primers representing each of the major classes of highly dispersed repetitive elements — B1, B2 and L1 — to amplify genomic fragments from inbred B6 mice (M. musculus) and M. spretus. Although the IRS-PCR patterns obtained upon agarose gel electrophoresis were extremely complex, it was possible to see clear evidence of species specificity. To simplify the patterns, these workers blotted and sequentially hybridized the IRS-PCR products to simple sequence oligonucleotides (12-mers containing three tandem copies of a tetramer) present frequently in the genome but only, by chance, in a subset of the amplified inter-repeat regions. The simplified patterns obtained allowed the identification and mapping of 13 new polymorphic loci.
Clearly, the utility of IRS-PCR as a general mapping tool is no greater than that of the RAPD technique or any other protocol that allows the random amplification of PCR fragments from around the genome. However, the real power of IRS-PCR is not in general mapping but in the identification and recovery of mouse-specific sequences from cell hybrids as discussed in Section 8.4.
Although the ability to identify and type simple basepair substitutions changed the face of genetics, it has not been a panacea. Finding RFLPs within a cloned region is often not easy; when they are found, their polymorphic content is often limited and di-allelic; finally, typing large numbers of RFLP loci by Southern blot analysis is relatively labor-intensive. Non-RFLP base changes can also be difficult to find, although this task has become easier with the development of the SSCP protocol. However, most SSCPs still show a limited polymorphic content with just two distinguishable alleles. Minisatellites are much more polymorphic than loci defined by nucleotide substitutions and minisatellite probes often allow one to simultaneously type multiple loci dispersed throughout the genome. However, minisatellite elements as a class have no unique sequence characteristics and are recognized only by the Southern blot patterns they produce when they are used as probes. The number of minisatellite loci uncovered to date numbers less than 1,000. Thus, in general, minisatellites cannot provide specific handles for typing newly cloned genes or genomic regions. Other methods of multi-locus analysis described previously suffer from the same limitations.
In the next chapter, it will be seen that one very important use of DNA markers is not to follow particular genes of interest in a segregation analysis, but rather to provide "anchors" that are spaced at uniform distances along each chromosome in the genome. Together, these anchor loci can be used to establish "framework maps" for new crosses which, in turn, can be used to rapidly map any new locus or mutation that is of real interest. If the number of anchors is sufficient, it will only take a single cross to provide a map position for the new locus. There is no need for anchor loci to represent actual genes. Their only purpose is to mark particular points along the DNA molecule in each of the chromosomes in a genome.
There are three criteria that define perfect anchor loci. First, they should be extremely polymorphic so that there is good chance that any two chromosome homologs in a species will carry different alleles. Second, they should be easy to identify so that one can develop an appropriate set of anchors for the analysis of any complex species that a geneticist wishes to study. Finally, they should be easy to type rapidly in large numbers of individuals.
Now with the dawn of the 1990s has come what may indeed be the magic bullet that geneticists (who study the mouse as well as all other mammals) have been waiting for — a genomic element with unusually high polymorphic content, that is present at high density throughout all mammalian genomes examined, is easily uncovered and quickly typed: the microsatellite. A microsatellite — also known as a simple sequence repeat (or SSR) — is a genomic element that consists of a mono-, di-, tri- or tetrameric sequence repeated multiple times in a tandem array.
Unlike other families of dispersed cross-hybridizing elements — such as B1, B2, and L1 — in which individual loci are derived by retrotransposition from common ancestral sequences (see Section 5.4), individual microsatellite loci are almost certainly derived de novo, through the chance occurrence of short simple sequence repeats that provide a template for unequal crossover events (as illustrated in Figure 8.4) that can lead to an increase in the number of repeats through stochastic processes. In general, microsatellite loci are not conserved across distant species lines, for example, from mice to humans, and it seems unlikely that these elements — which are practically devoid in information content — have any functionality either to the benefit of the host genome 64 or in and of themselves. Microsatellites do not appear to be selfish elements (discussed in Section 5.4). Rather, microsatellites, like minisatellites, are simply genomic quirks that result from errors in recombination or replication.
Microsatellites containing all nucleotide combinations have been identified. However, the class found most often in the mouse genome contains a (CA)n•(GT)n dimer, and is often referred to as a CA repeat. The existence of CA repeats, their presence at high copy number, and their dispersion throughout the genomes of a variety of higher eukaryotic species was first demonstrated a decade ago by several different laboratories (Miesfeld et al., 1981; Hamada et al., 1982; Jeang and Hayward, 1983). Although independent examples of CA-repeat polymorphisms surfaced over the following decade, it was not until 1989 that three groups working independently uncovered sufficient evidence to suggest that microsatellites as a class were intrinsically extremely polymorphic (Pickford, 1989; Weber and May, 1989; Pedersen et al., 1993). Further systematic studies have confirmed the high level of polymorphism associated with many microsatellite loci in all higher eukaryotes that have been looked at.
Without PCR, most microsatellites would be useless as genetic markers. Allelic variation 65 is based entirely on differences in the number of repeats present in a tandem array rather than specific basepair changes. Thus, the only way in which alleles can be distinguished is by measuring the total length of the microsatellite. This is most readily accomplished through PCR amplification of the microsatellite itself along with a small amount of defined flanking sequence on each side followed by gel electrophoresis to determine the relative size of the product as illustrated in Figure 8.10.
Microsatellite loci can be identified in two ways — by searching through DNA sequence databases or by hybridization to libraries or clones with an appropriate oligonucleotide such as (CA)15. In the former case, flanking sequence information is obtained directly from the database. In the latter case, it is first necessary to sequence across the repeat region to derive flanking sequence information. A unique oligonucleotide on each side of the repeat is chosen for the production of a primer according to the criteria described in the front of Section 8.3. It is best to choose two primers that are as close to the repeat sequence as possible — the smaller the PCR product, the easier it is to detect any absolute difference in size. Variations in the length of PCR products can be detected by separation on NuSieve™ agarose (FMC Corp.) gels (Love et al., 1990; Cornall et al., 1991) or polyacrylamide gels (Weber and May, 1989; Love et al., 1990). Agarose gels are easier to handle, but polyacrylamide gels provide higher resolution. When alleles are difficult to resolve with native gels, it is often possible to improve the level of resolution by running denaturing gels. Bands are detected by ethidium bromide or silver staining of gels or by autoradiography of PCR products formed with labeled primers.
An even higher level of cost efficiency can be achieved by combining two or more loci for simultaneous analysis through multiplex PCR. Samples can be combined before the PCR reaction — if the different primer pairs have been shown not to cause combinatorial artifacts — or after the PCR reaction but before the gel run. In all cases, the entire process is amenable to automation.
Microsatellites can be classified first according to the number of nucleotides in the repeat unit. Mononucleotide and dinucleotide repeat elements are quite common; with each subsequent increment in nucleotide length — from trinucleotide to tetranucleotide to pentanucleotide — the frequency of occurrence drops quickly. Perfect microsatellites are those that contain a single uninterrupted repeat element flanked on both sides by non-repeated sequences (Weber, 1990). A large proportion of microsatellite loci are imperfect with two or more runs of the same repeat unit interrupted by short stretches of other sequences. The polymorphic properties of imperfect microsatellites are determined by the longest stretch of perfect repeat within the locus. Not infrequently, microsatellites are of an imperfect and compound nature, with a mingling of two or more distinct runs of different repeat units.
The most common microsatellites in the mouse genome are members of the dinucleotide class. With complementarity and frame-shift symmetry, there are only four unrelated types of dinucleotide repeats that can be formed:
Based on a quantitative dot blot analysis, Hamada and his colleagues (1982a, 1982b) estimated the number of CA repeat loci in the mouse genome at ~100,000, equivalent to an average of one locus every 30 kb. Another estimate of CA-repeat copy number was obtained by scanning 287 kb of mouse genomic sequences entered in GenBank for (CA)n where n was six or greater (Stallings et al., 1991). This analysis found CA-repeats once every 18 kb on average. The difference between these two estimates can be accounted for entirely by sequences having only 6-9 repeats, which are too short to be detected by the hybridization-based dot blot analysis (Weber, 1990).
The second most frequent microsatellite class in the mouse genome is the GA repeat, which occurs at a frequency of approximately half that observed for CA repeats (Cornall et al., 1991). GA-repeat loci are just as likely to be polymorphic as CA-repeat loci. Thus by screening for both simultaneously, one can increase the chances of finding a useful microsatellite by 50%.
The mononucleotide repeat poly(A•T) is found in the mouse genome at a frequency similar to, if not greater than, the CA repeats. However, it is often contained within the highly dispersed B1, B2, and L1 repeats, which are themselves present in ~100,000 copies per haploid genome. Thus, random screens for poly(A•T) tracts will frequently land investigators in these more extensive repetitive regions where it will be difficult to derive locus-specific primer pairs for PCR analysis. Nevertheless, if one is aware of this pitfall, it becomes possible to use computer programs to assist one in this task, and it is often possible to type microsatellite-containing B1 (or B2 or L1) elements (Aitman et al., 1991). Mononucleotide repeats, both within and apart from the more complex repeat elements, are just as likely to be polymorphic as dinucleotide repeats (Aitman et al., 1991). However, a second potential pitfall with long poly(A•T) tracts is that, as is the case with long (TA)n•(AT)n dinucleotide tracts, there is a reduced melting temperature which necessitates the use of PCR elongation steps under conditions of reduced specificity, leading to an increased incidence of artifactual products. As a consequence of these pitfalls, poly(A•T) tracts have been used much less frequently as a source of polymorphic microsatellite markers. The microsatellite poly(C•G) is not associated with either of these pitfalls, but it is much less frequently observed — by an order of magnitude — in the mouse genome (Aitman et al., 1991).
Tri- and tetranucleotide repeat unit microsatellites are also present in the genome, but at a frequency ten-fold below that of the dinucleotide (CA)n and (GA)n loci (Hearne et al., 1992). 67 As such, they will be represented much less often in genomic libraries and individual clones. However, once uncovered, these higher-order microsatellites are much better to work with than the dinucleotide loci. The level of polymorphism observed with the tri- and tetranucleotide loci appears similar to that observed with CA and GA repeats, but alleles are much more readily resolved with 3-4 bp mobility shifts for each repeat unit difference. Furthermore, ladders of artifactual PCR products commonly seen with dinucleotide repeats do not appear as often or as intensely with higher-order repeat unit loci (Hearne et al., 1992).
As is the case with minisatellite loci, the generation of new microsatellite alleles is not due to classical mechanisms of mutagenesis. Rather, the number of tandem repeats is altered as a consequence of mispairing, or slippage, during recombination or replication within the tandem repeat sequence. As illustrated in Figure 8.4, events of this type will create new alleles by expanding or contracting the size of the locus. The frequency with which these events occur is a function of the number of repeats in the locus with a sigmoidal distribution. CA-microsatellites with 10 or fewer repeat units are unlikely to show polymorphism; with 11 to 14 repeat units, there is an intermediate, and climbing, probability of detecting polymorphism; with 15 repeat units or more, there is a maximal probability of detecting polymorphism (Weber, 1990; Dietrich et al., 1992). Thus, to maximize the probability of detecting polymorphism, one should focus analyses on CA-repeat loci having n >= 15. Hybridization screens can be set up to accomplish this task by probing blots with a (CA)15 oligonucleotide under high stringency conditions of 65°C with 0.1 X SSC (Dietrich et al., 1992).
A large number of laboratories have now reported the results of investigations into the frequencies at which microsatellite polymorphisms are detected in comparisons of two or more inbred strains or mouse species. The actual results would be expected to vary depending on the method used to recover microsatellites (because this will determine the lower boundary for repeat number) and the method used to type the PCR products (because agarose gels are less resolving than polyacrylamide gels). In an analysis of over 300 CA-repeat microsatellites that are predominantly of the n >= 15 class, an average polymorphism rate of approximately 50% was observed in pairwise comparisons among nine classical M. musculus inbred strains; the lowest level of polymorphism observed was 35% between DBA/2J and C3H/HeJ, and the highest was 57% between B6 and LP/J (Dietrich et al., 1992). Not unexpectedly, even higher levels of polymorphism were observed in pairwise comparisons between classical inbred strains and other Mus species or subspecies. The rate of polymorphism between B6 and M. m. castaneus was 77%, and between B6 and M. spretus, it was ~90% (Love et al., 1990; Dietrich et al., 1992). For a small but significant number of loci, the primers designed to amplify an inbred strain locus failed to amplify an allelic product from the M. spretus genome (Love et al., 1990); this is almost certainly due to an interspecific polymorphism in a target sequence recognized by one of the flanking primers.
A number of investigators have attempted to measure the rate at which new microsatellite alleles are created. This can be readily accomplished in the mouse where the relationships among a large number of different inbred strains have been well documented and it is possible to count the generations that separate various strains from each other (Bailey, 1978). The results of these studies indicate that the rate of mutation is highly variable — over at least an order of magnitude. This variability could be a consequence of genomic position effects, but the mechanism of allele generation must be clarified before one can say for sure. In the most comprehensive analysis to date, Dietrich and colleagues (1992) analyzed the average rate of mutation at 300 loci within the BXD set of recombinant inbred strains. The average mutation rate was calculated at one in 22,000 per locus per generation, which is 5-50-fold greater than that normally attributed to mutagenesis at classical loci. This average microsatellite "mutation" rate is high enough to allow the generation of a large amount of polymorphism among individuals within a species, but low enough to allow one to accurately follow the segregation of two or more alleles from one generation to the next within a typical genetic cross.
The high level of polymorphism associated with microsatellites (as a class) represents just one component of their rapid rise to become the "genetic tool of choice" for mappers working with all animal species. Their uniqueness and power also lies within the ease with which they can be uncovered, the ease with which they can be typed, and the ease with which they can be disseminated. To develop a panel of microsatellite loci for analysis of the mouse genome, Todd and his colleagues simply searched through the EMBL and GenBank databases for entries that contained (CA)10, (GA)10, or their complements (Love et al., 1990). To increase the size of this panel for higher resolution mapping analysis, genomic libraries constructed to contain short inserts were screened with CA-repeat probes, and positive clones were isolated and sequenced (Cornall et al., 1991; Dietrich et al., 1992).
Using a combined panel of 317 microsatellite loci, the Whitehead/MIT Genome Center developed a first-generation whole mouse genome linkage map with an average spacing of 4.3 cM (Dietrich et al., 1992). With the publication of the oligonucleotide sequences that define and allow the typing of each locus, the markers became available to everyone in a democratic fashion. As of January, 1994, the Whitehead group had defined and mapped over 3,000 microsatellite loci. 68 Up to date mapping, strain distribution, and sequence information on all of these loci can be obtained electronically as described in Appendix B. Furthermore, the commercial concern Research Genetics Inc. has made life even easier for the mouse genetics community by offering each primer pair in this panel at a greatly reduced cost relative to custom DNA synthesis.
Since microsatellite typing is PCR based, and there is usually no need for blotting or probing, results can be obtained rapidly with a minimal expenditure of often precious material and always precious man- and woman-hours. Dietrich and colleagues (1992) reported that two scientists can "genotype new crosses for the entire genome in a few weeks per cross" which represents an order of magnitude improvement over RFLP-based approaches.
Microsatellites can serve not only as tags for anonymous loci but for functional genes as well. Stallings and his colleagues (1991) found that 78% of the clones from a mouse cosmid library have CA repeats. If one also searched for GA repeats, the percentage of microsatellite-positive cosmid clones would be even greater. An even higher probability of identifying microsatellite loci — close to 100% — can be achieved with clones recovered from larger insert libraries constructed with yeast artificial chromosomes (YACs) or special prokaryotic vectors (see Section 10.3.3). Small fragments that contain the microsatellite can be subcloned and sequenced to identify a unique set of flanking primers for genetic analysis. Microsatellites can truly be viewed as universal genetic mapping reagents.
During the 1980s, the difficulties encountered in the search for RFLPs among the classical inbred strains led to the emergence of the interspecific cross — between a M. musculus-derived inbred strain and M. spretus — which became a critical tool for the development of the first high-resolution DNA-locus-based maps of the mouse genome (Avner et al., 1988; Copeland and Jenkins, 1991 and Section 9.3). Interspecific backcross panels still represent a powerful tool for mapping newly characterized DNA clones. However, with microsatellites, it is now possible to go back to classical crosses among M. musculus strains to map interesting phenotypic variants as discussed in Section 9.4.