Mutations at the T/t complex in the mouse have been under study now for just over 50 years, and have proved valuable tools for analyzing such diverse situations as embryogenesis, spermatogenesis, and population dynamics in wild mice.
The first mutation at this region of chromosome 17 was reported in 1927 by Dobrovolskaia-Zavadskaia (
1). She described a dominant gene,
T
that resulted in heterozygotes that were short-tailed, and homozygotes that died during prenatal development. Subsequent breeding of
T-bearing
mice to apparently normal animals that were (significantly for the rest of this story) in at least one case derived from wild-caught
mice revealed the presence of recessive (
t)
mutations. These genetic variants interacted with the dominant
T
to produce a new phenotype, complete taillessness (
2). These first recessive mutations detected were also lethal when homozygous and behaved as genetic alleles to
T;
thus tailless lines of the same ancestry bred as balanced lethal systems:
| T/t | x | T/t | ||
| | | V |
||||
| T/T | T/t | t/t | ||
| (dead as embryos) |
tailless | (dead as embryos) |
L.C. Dunn and his colleagues at Columbia took up the study of the T/t system in the 1930s and soon showed that these mutations differed from conventional genes in a number of properties that are outlined below.
The genetic analysis of the first tailless stocks studied showed that lethal t-factors of independent origin could differ from one another sufficiently to show a degree of genetic complementation. The test for this was simple and straightforward; matings of tailless mice from different lines did not behave as balanced lethal crosses, but rather produced two classes of offspring as diagrammed below for the original cross that defined this phenomenon ( 3).
| line A (T/t0) | x | line 29 (T/t1) | ||||
| | | V |
||||||
| T/T | T/t0 | T/t1 | t0/t1 | |||
| (dead as embryos) |
tailless | viable normal-tailed |
||||
Complementation is generally not perfect, and tx/ty compounds are often both morphologically abnormal and subviable. At the present writing, we have studied more than 50 recessive lethal t-mutations of independent origin. By complementation analysis we found them to fall into six complementation groups as follows:
| t0 | ( t1, t6, t30, tw4) |
| t9 | ( t4, tw18, tw30, tw52) |
| t12 | ( tw32) |
| tw1 | ( tw3, tw12, tw20, tw21, tw71, tw72) |
| tw5 | ( tw6, tw10, tw11, tw13, tw14, tw15, tw16, tw17, tw38, tw39, tw41, tw46, tw47, t74, tw75, tw80, tw81, tw93, tw94, tw97) |
| tw73 |
One of the most remarkable effects of t-mutations is an abrogation of Mendel's rules in males. This departure from conventional genetics became obvious early in the study of the T/t complex, when animals of the genotype T/ t0 were outcrossed to normal (+/+) mates. Tailless females produced the expected proportions of 50% short-tailed (T/+) and 50% normal tailed (T0/+) offspring, but tailless males transmitted T0 to a surprisingly high degree; about 80% of the offspring were normal tailed. Tests of t0/+ males mated to T/t females showed that the effect, called transmission ratio distortion, was directly attributable to the t0 factor, not to any interaction with T ( 3). All evidence since that time has continued to demonstrate that transmission ratio distortion is a general feature of lethal and semilethal t-factors (the single exception being the T9 complementation group). A number of cases have been described of specific t-mutations with transmission ratios of the lethal gene that are over 95%, so it is clear that this situation represents an extreme departure from conventional genetic systems. The mechanism of this distortion does not apparently depend on meiotic disturbance nor on degeneration of one type of sperm, nor on such factors as embryo selection. The best guess at the moment is that t-bearing sperm have some superiority in fertilizing ability that is conferred by haploid gene function after meiosis ( reference 4 for review). Other effects of t-factors in males have also been described; both homozygotes for semilethal t's and males carrying two different lethal complementing (tx/ty) mutations are completely sterile. In both these cases, pronounced morphological abnormalities occur in developing spermatids ( 5, 6), so the basis for sterility appears to be quite different from that of distorted transmission ratio. Nevertheless, the two phenomena are presumably related in some way.
Both lethal and semilethal t-factors in heterozygous condition suppress recombination over a long stretch of chromosome. The recombination suppression effect may vary slightly from t-haplotype to t-haplotype, but in general effectively includes the distance from the locus of T to, but not much beyond, the H-2 region ( 7). Although this obviously leads to suggestions that t-mutations represent inversions or deletions of some magnitude, numerous high resolution cytological studies have failed to provide any evidence for chromosome aberrations ( reference 4 for review).
The initial studies of the genetic behavior of t-mutations suggested that they were "hypermutable," since they frequently (about 1/500 gametes) produced what Dunn and his colleagues referred to as "exceptions." Exceptions were detected in balanced lethal crosses by the appearance, usually singly, of normal-tailed offspring, which proved on genetic analysis to carry two different complementing t-factors ( 8). Once chromosome markers became available and were incorporated into t-bearing stocks, it became apparent that the great majority of such exceptions were accompanied by recombination. Most studies have made use of the marker tf ( tufted), thus:
| T tf | x | T tf | ||||
| tn + | tn + | |||||
| | | V |
||||||
| T tf | T tf | tn + | ||||
| T tf | tn + | tn + | ||||
| lethal | tailless | lethal | ||||
| and ! | ||||||
| tn + | ||||||
| t! tf | ||||||
viable
normal-tailed
The majority of exceptional chromosomes generated by lethal or semilethal t-factors have proved to be viable when homozygous, and in fact to retain as their only t-like property the ability to interact with T to produce taillessness. No revertants to complete wild-type have ever been detected, however. Over 100 exceptions have been analyzed, and of these 96 were viable and only nine lethal. All of the viable t-factors generated in stocks marked with tf were recombinants. These data lead quite inescapably to the conclusion that lethal and semilethal t-haplotypes are at the very least bilocal and consist of a minimum of two separable elements, one that is allelic with T and responsible for interaction with it to produce taillessness, and one near tf that is involved with lethality, recombination suppression, and effects on sperm function. Detailed analysis of recombination-derived t-factors suggests even more complicated models are possible ( 9), but these are sufficiently speculative not to warrant discussion in the present context.
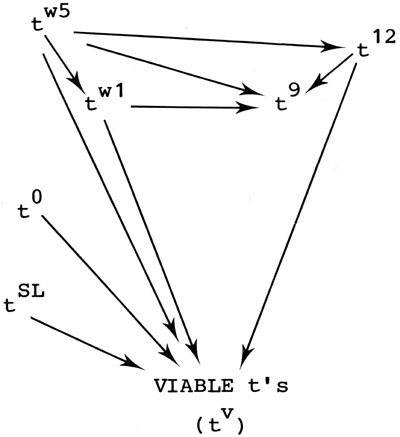
Relatively few lethal t-mutations have been derived as exceptions from pre-existing factors. No doubt many that do arise are lost before birth because of poor complementation with the parent allele, and others that occur in male tn/t! compounds are lost to analysis because of the inevitable sterility associated with that genotype. For these reasons it is virtually impossible to get reliable estimates of the spectrum of frequency of newly generated lethal t's. Nevertheless, those that have been analyzed show an interesting unidirectional pattern of occurrence. As seen in the diagram below, tw5 has never been generated from a preexisting t, but has given rise to virtually all others. Likewise, t9, which has never itself given rise to a lethal, can be produced by most lethals.
The event by which one lethal t factor converts to a different one is not well defined. Only one lethal ( tw32) has been generated in stocks marked with tf where recombination could be assayed. In this one case, the origin of Tw32 was clearly not accompanied by crossing-over between T and tf ( 10). This suggests that viables and lethals may originate by different mechanisms ( 11), with lethals perhaps arising by mutation rather than recombination. It is worth emphasizing here that no instance of mutation from a wild-type gene to a t-factor of any sort has ever been observed; all t-mutations studied have either been derived from wild populations or obtained as exceptions from pre-existing t-factors.
The first exploration for t-mutations in wild populations of mice was apparently initiated because L.C. Dunn saw a possible correlation among three factors: the original detection of t-mutations by Dobrovalskaia-Zavadskaia in the descendents of wild mice, the high transmission ratio of t-factors through males, and their apparent hypermutability in the laboratory. He obtained representatives of a closed colony of mice originated by Schneider ( 12) from animals caught wild in suburbs of Philadelphia and New York. In the original study 10 males were tested for the possible presence of a t-mutation by mating to T/+ females. Of the eight which actually bred, five tested positive for the presence of t-factor by producing tailless offspring. The transmission ratio of the t-mutation(s) found in this population was, as predicted, high (86%) ( 13).
This sample was only the first of many in which t-factors were found in wild populations of mice. Their distribution is ubiquitous, and t-factors have been found all over the world: North America, Europe, Australia, Asia, and South America.
To date, as summarized by Klein ( 14) with additions of unpublished data from Dunn and Bennett, 52 separate populations have been sampled for their content of t-factors; 27 were found to contain t-factors and 31 were negative. The proportion of negative populations is undoubtedly artificially high, because of the small sample size available for most of them; generally under 10 and more often 2-4 animals were tested. Actually only two of the negative populations can be considered as subjected to a valid test. A sample of 23 animals from a farm in Calgary, Canada, contained no t-factor, although other nearby farms had mice that did ( 15). Likewise, 40 animals from Great Gull Island in Long Island Sound were analyzed over a period of several years without detecting a t-factor and that population can be securely considered to be free of t-mutations ( 16). The fact that this was an island population may be a significant factor (see below). Table 1 gives information on sample sizes, numbers of animals tested, and gene frequency. All of the t-haplotypes found in these wild populations were lethal and semilethal, but the distribution of complementation groups was very unequal; some populations contained more than one complementation group. Table 2 shows that almost twice as many examples of the tw5 group were found than all others combined, while semilethal haplotypes and representatives of the tw1 complementation group comprised most of the other t-mutations that were defined. One member of the t0 group and the only example known of tw73 were also found. Neither t12 nor t9 were represented in these samples. Again, every t-haplotype found proved to have a high male transmission ratio, always over 80% and not infrequently in the 95-98% range.
Tested populations that did contain a t-factor had an overall heterozygote frequency of 0.35, which is of course extraordinarily high for a lethal or semilethal gene. Even if the assumption is made that most so-called "negative" populations were insufficiently tested, and these data (with the exceptions of the two quite securely negative populations, see a in Table 1) are pooled with those from positive populations, an estimated heterozygote frequency of 0.25 is obtained for mouse populations in general. Thus, the conclusion must be that lethal or semilethal t-haplotypes are a natural polymorphism in wild mice, with gene frequencies of 10% or more.
This situation is not completely without precedent since, for example, the gene for sickle cell anemia, which is virtually a homozygous lethal, has comparable gene frequencies in some human populations, where it appears to be maintained by the advantage conferred on heterozygotes in respect to protection from malarial parasitism ( 17). Although no strong evidence has been found for any conventional relative advantage in fitness in t-heterozygotes ( 18), an obvious heterozygous advantage, although of a rather unique sort, occurs in t-heterozygotes due to male transmission ratio distortion.
There have been several attempts to analyze in theoretical terms the apparent ability of transmission ratio distortion to maintain a polymorphic frequency of lethal or semilethal genes in wild populations of mice, and most have been instructive in various ways. The first formally valid approach was proposed by Bruck ( 19) who generated a deterministic model based on the Hardy-Weinberg parameters of infinite population size and random mating. His equation took into account the necessary factors of embryonic lethality of homozygotes and segregation distortion in males only, but led to a prediction that lethal t-gene frequencies at equilibrium would be far higher (between 30 and almost 50%) than those actually found in nature (about 10%). A somewhat more sophisticated version that took semilethality into account was later produced by Dunn and Levene ( 20) and suffered from the same inability to predict the gene frequencies in natural populations. The failure of these deterministic solutions to correspond to the real world was actually very informative, because it suggested that mouse populations probably were subject to stochastic rules that were imposed because of small effective population sizes where random genetic drift would be an important force. Actually, there were already data being collected, that are now much more extensive ( reference 15 for review), to show that mouse breeding populations are divided into extremely small subunits called demes. The average deme may consist of about 8 or 10 mice, usually not more than 12, and is relatively isolated from other groups since demes are usually governed by a dominant male who excludes intruders. The isolation of demes is not fixed or permanent, however, since the absence or death of the dominant male is likely to be quickly followed by the immigration of another.
The suggestion that effective breeding populations are indeed small was bolstered very much by stochastic models proposed by Lewontin and Dunn ( 21) for the behavior of t-factors in wild mice. These interesting studies involved computer simulations of small breeding populations of mice, which were set up with designated numbers of parents of desired genotypes and transmission ratio; a "Monte Carlo" method of gamete selection from the resulting gene pool was used to generate the next generation, and so on. One of the most telling points that emerged from this study was that, under these conditions population sizes of 25 males and 25 females, or even 10 males and 10 females, behaved in a manner that approached Hardy-Weinberg predictions of a t-gene frequency of about 40%. Thus, for the mouse, 20 appears to represent an infinite population size! When the simulated populations were reduced to a size of about eight, it became apparent that any given population was virtually certain, given enough time, to lose its t-factor and reach fixation for the wild type. The length of time a population maintained a t-haplotype was heavily dependent on the value set for transmission ratio, with only very high ratio factors persisting for appreciable lengths of time. On the other hand, it also appeared that a single male with a high ratio factor could be highly effective in introducing that factor into an otherwise negative deme. Taken together these observations suggested that mouse populations as a whole were divided into small demes, each of which, if it had a t-factor in it, was nevertheless progressing ultimately to fixation of the wild type alternate, but each of which, if free of t-factors at the moment, was subject to a certain probability of "re-infection" by the immigration of a heterozygous male. In this context it is significant that one of the two securely t-free populations so far discovered was the Great Gull Island population, which exists on an essentially uninhabited island, where inward migration would be very restricted, although not impossible since boats do visit the island. Anderson et al. ( 22) and Bennett and associates ( 23) used this population as a kind of natural laboratory to study the effects of introducing males heterozygous for a wild-derived lethal t-haplotype. In 1956 and 1957, +/ tw11 males were released in one small area near the center of the island, and samples taken yearly between 1959 and 1962 and again in 1966 showed not only that tw11 had persisted in the population, but that it had spread virtually throughout the island ( Figure 1). However, in 1969, 1970, and 1972, additional samples studied were free of t-mutations. The total number of animals sampled was large enough ( 39) to suggest strongly that complete fixation of the wild type had been reached. Thus, it is likely that the natural population on Gull Island fulfilled the predictions of the stochastic models of Lewontin and Dunn for 1) easy infection by t/+ heterozygous males and 2) eventual loss from an isolated population. A definitive solution of this point has unfortunately not been possible, because in 1974 representatives of the American Museum of Natural History in New York, who supervise the island and study populations of terns that nest there, permitted the escape of pet-store mice into the natural population and thus rendered it useless for our purposes.
Stochastic models discussed above led to gene frequency predictions that were relatively low and far more compatible with data from actual wild populations than were the predictions of deterministic models. In general terms, the actual and theoretical picture that emerges is that most populations of mice carry t-mutations in some substantial frequency. The question then arises whether this polymorphism for lethal or semilethal genes confers some intrinsic adaptive advantage to mouse populations and, if so, what the advantage is.
Perhaps the most obvious possibility is some relationship with the H-2 complex. The observation that only lethal and semilethal t-factors have high transmission ratios and are found in wild populations, and significantly that only these inhibit recombination through the region of H-2, has led to speculations ( 24, 25, 26) that the T/t complex and the H-2 complex may have some functional or evolutionary relationship.
Evidence for an actual physical relationship has indeed been found. Hammerberg and Klein ( 7) and Hammerberg et al. ( 27) have serologically examined H-2 types in t-bearing chromosomes and found only a restricted number of H-2 types in that sample. With a few exceptions, t-haplotypes of the same complementation group, no matter how disparate their geographical origin, had identical H-2 haplotypes. These findings have been confirmed with respect to I-region genes by McDevitt et al. ( 28). These findings are particularly surprising in view of the extensive polymorphism of H-2 in wild mouse populations ( 29), which makes it highly unlikely that mice from separate populations would carry the same H-2 region genes. Obviously, this instance of linkage disequilibrium could either have a functional or evolutionary interpretation. It could be assumed that particular t-haplotypes form especially advantageous gene complexes (super-genes?) with particular H-2 haplotypes, and thus are selected for in wild populations. Alternatively, it could be assumed that t-haplotypes of the same complementation group must often actually represent the same chromosome which originated once and then was dispersed geographically by migration, with the t-haplotype and the existing H-2 complex held permanently together by recombination suppression. This is perhaps the most likely explanation, since it implies that the "primordial" mutation to a t-factor is a very rare event; this seems indeed to be the case since none have been observed in the laboratory. The rare instances of lethal or semilethal t-haplotypes with "inappropriate" H-2 partners are also explainable on this hypothesis, since at present it appears that the generation of one lethal from another is not only a rare event but one that is not recombinational but mutational in nature. Thus a new lethal generated by mutation would necessarily remain associated with an H-2 type typical of the parent complementation group.
In summary, at present it is clear that the lethal and semilethal t-factors that suppress recombination at least as far as the H-2 region and have high male transmission ratios are polymorphic in natural populations of mice. Whether they confer an adaptive advantage is not yet clear, but if they do, it is likely to be due to some as yet undefined relationship to H-2 region genes.
1. Dobrovolskaia-Zavadskaia, N. (1927). Comp. Rend Soc. Biol. (Paris) 97: 114.
See also
MGI.
2. Dobrovolskaia-Zavadskaia, N., and Kobozieff, N. (1932). Comp. Rend. Soc. Biol. (Paris) 110: 782.
3. Chesley, P., and Dunn, L.C. (1936). Genetics 21: 525.
4. Bennett, D. (1975). Cell 6: 545.
5. Dooher, G.B., and Bennett, D. (1974). J. Embryol. Exp. Morphol. 43: 749.
See also
PubMed.
6. Dooher, G.B., and Bennett, D. (1977). Biology of Reproduction 17: 269.
See also
PubMed.
7. Hammerberg, C., and Klein, J. (1975). Nature 258: 296.
See also
PubMed.
8. Dunn, L.C., and Gluecksohn-Schoenheimer, S. (1950). Proc Natl. Acad. Sci. USA 36: 233.
9. Lyon, M.F., and Mason, I. (1977). Genet. Res. 29: 255.
10. Dunn, L.C., Bennett, D., and Beasely, A.B. (1962). Genetics 47: 285.
See also
MGI.
11. Klein, J., and Hammerberg, C. (1977). Immunol. Rev. 33: 70.
See also
MGI.
12. Schneider, H.A. (1946). Proc. Soc. Exp. Biol. Med. 63: 161.
13. Dunn, L.C., and Morgan, W.C., Jr. (1952). Amer. Nat. 536: 321.
14. Klein, J. (1975). Biology of the Mouse Histocompatibility-2 Complex Springer-Verlag, New York.
See also
MGI.
15. Anderson, P.K. (1964). Science 145: 177.
See also
PubMed.
16. Dunn, L.C., Beaseley, A.B., and Tinker, H. (1960). J. Mammal. 41: 220.
See also
MGI.
17. Allison, A.C. (1954). Brit. Med. J. 1: 290.
See also
PubMed.
18. Dunn, L.C., Beasley, A.B., and Tinker, H. (1958). Amer. Nat. 92: 215.
19. Bruck, D. (1957). Proc. Natl. Acad. Sci. USA 43: 152.
20. Dunn, L.C., and Levine, H. (1961). Evolution 15: 385.
21. Lewontin, R.C., and Dunn, L.C. (1960). Genetics 45: 795.
22. Anderson, P.K., Dunn, L.C., and Beasley, A.B. (1964). Amer. Nat. 98: 57.
23. Bennett, D., Bruck, D., Dunn, L.C., Klyde, B., Shutsky, F., and Smith, L.J. (1967). Amer. Nat. 101: 538.
24. Snell, G.D. (1968). Folia Biol. (Praha) 14: 335.
See also
PubMed.
25. Klein, J. (1975). Adv. Exp. Med. Biol. 64: 467.
See also
PubMed.
26. Artzt, K., and Bennett, D. (1975). Nature 256: 545.
See also
PubMed.
27. Hammerberg, C., Klein, J., Artzt, K., and Bennett, D. (1976). Transplantation 21: 199.
See also
PubMed.
28. McDevitt, H.O., and Levenson, J.R. (1976). J. Exp. Med. 144: 834.
See also
PubMed.
29. Klein, J. (1974). Ann. Rev. Genet. 8: 63.